Амиотрофия спинальная прогрессирующая верднига гоффмана. Типы сма. Спинальная амиотрофия и ее формы
Частота заболевания
СМА – одно из самых часто встречеющихся заболеваний из орфанных (редких), болеет один новорожденный на 6000-10000 .
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
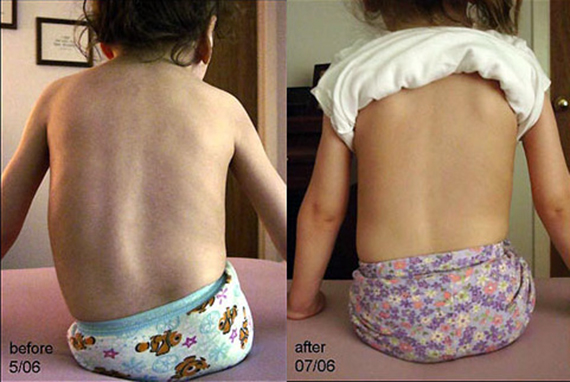
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.

От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III , болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА
IV
, этот тип называется ещё «взрослой СМА»,
поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300 .
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?

Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Итальянка Симона Спиноглио
родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен — о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов . Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий — член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Спинальная амиотрофия Верднига-Гоффмана представляет собой наследственное заболевание. На фоне патологии отмечается повреждение в двигательных нейронах. Спинальная амиотрофия Гофмана передается с неполовыми хромосомами. Далее подробнее рассмотрим заболевание, его клиническую картину и возможные терапевтические мероприятия для его устранения.
Терминология
Прежде чем говорить о том, как проявляет себя спинальная амиотрофия, ознакомимся с некоторыми понятиями. Разберем название патологии. Оно состоит из двух частей:
- Спинальная - слово указывает на локализацию нарушения. В данном случае речь идет об определенном элементе, находящемся в позвоночнике. Это одна из важнейших структур организма - спинной мозг.
- Амиотрофия - слово, включающее три части: "а" - нарушения, "мио" - мышца" и "трофия" - питание.
На основании этой информации можно понять значение названия патологии. Спинальная амиотрофия Верднига-Гофмана, таким образом, представляет собой нарушение питания в мускулатуре. Патология характеризуется наличием слабости и подергиванием волокон.
Наследование
Спинальная мышечная амиотрофия относится к аутосомно-рецессивным заболеваниям. Это определение указывает на тип наследования, при котором передача признака осуществляется посредством неполовых хромосом. При этом он проявляется только тогда, когда присутствует изначально у обоих родителей (сами они могут не болеть).

Развитие заболевания
Спинальная амиотрофия взрослых не встречается. Патология проявляет себя у детей. Недуг характеризуется злокачественным течением и быстрым прогрессированием. За координацию движений отвечают крупные клетки спинного мозга. Они также поддерживают тонус мускулатуры. При их повреждении развивается дисфункция мышц.
Врожденная форма
Спинальная амиотрофия имеет три формы. Они определяются в соответствии со временем проявления первых признаков и интенсивностью развития процесса. Врожденная форма может начаться еще во внутриутробном периоде. В данном случае отмечается ослабление шевеления плода на более поздних сроках беременности. При этом в начале дородового периода движения были в пределах нормы. Само разрешение беременности может оказаться патологическим. Зачастую уже в течение первых нескольких дней после рождения обнаруживаются выраженные парезы мускулатуры, сопровождающиеся снижением ее тонуса и ухудшением сухожильных рефлексов. Также могут отмечаться ретробульбарные (ранние) симптомы. Они проявляются слабым криком младенца и вялым сосанием. В ряде случаев наблюдается полная арефлексия. У ребенка могут быть выявлены фибрилляции в языке, гипомимия, понижение глотательного рефлекса. Спинальная амиотрофия сопровождается тахикардией. Зачастую патология сочетается с несколькими пороками развития, замедлением формирования психики. Спинальная амиотрофия отличается быстрым течением и заканчивается к 1-1,5 годам летальным исходом.

Ранняя форма
Она отличается более мягким течением, нежели врожденная. Ранняя детская форма считается классическим проявлением заболевания. Спинальная амиотрофия в этом случае проявляется в возрасте до полутора лет.
Практически во всех случаях признаки недуга обнаруживаются после пищевого отравления или какого-либо инфекционного поражения. Развивающийся нормально ребенок начинает достаточно быстро терять ранее приобретенные двигательные способности. Он перестает сидеть, стоять и ходить. Сначала отмечаются вялые парезы в нижних конечностях, постепенно переходящие на туловище и руки. Состояние ребенка ухудшается очень быстро. В мышцах шеи и бульбарной мускулатуре появляется слабость. В результате недостаточности дыхательной системы к 4-5 годам появляется пневмония, затем наступает смерть. Вялые парезы у детей осложняются сухожильными контрактурами. Зачастую спинальная амиотрофия Верднига-Гоффмана сопровождается общим гипергидрозом.
Позднее появление патологии
Третья форма заболевания начинается после 1,5-2 лет. По сравнению с предыдущими она протекает сравнительно легко. Способность к передвижению сохраняется у детей до 10 лет. После этого состояние, как правило, ухудшается.

Клиническая картина
Для патологии характерны парезы сначала проксимальных отделов нижних конечностей, а затем и верхних. При спинальной амиотрофии жировой подкожный слой хорошо выражен. Это, в свою очередь, затрудняет выявление дисфункции мускулатуры. Достаточно рано начинают угасать сухожильные рефлексы. Для патологии характерным является малый тремор пальцев при вытянутых руках. Типичными считаются деформации костей, в особенности нижних конечностей и грудины. Бульбарные симптомы проявляются в виде атрофии мускулатуры языка с подергиваниями фибриллярного типа, парезом в мягком небе и сниженным глоточным рефлексом.
Болезнь Фацио-Лонде
Это особый вариант проявления атрофии. Патология начинает развиваться, как правило, к трем годам жизни, и в некоторых случаях в подростковом возрасте. Для недуга характерна слабость мышц лица, в том числе жевательной мускулатуры. Отмечается затруднение глотания и изменения голоса. Патология сопровождается атрофией языка, в некоторых случаях может появиться офтальмоплегия. Прогрессирует болезнь очень быстро. Спустя 6-12 месяцев наступает смерть. К бульбарным нарушениям могут добавиться параличи и парезы в конечностях. В некоторых случаях эти симптомы не успевают даже развиться. Тем не менее, при вскрытии всегда выявляется поражение в клетках передних спинномозговых рогов на всем протяжении.

Диагностика
При проведении обследования патологию отделяют от миотонии Оппенгейма. Большинство специалистов считают, что эта патология не является самостоятельной нозологической единицей. Миотония Оппенгейма, по мнению исследователей, -синдром, для которого ведущим проявлением становится гипотония мышц выраженного типа. В этой связи в последнее время широко распространен термин "вялый ребенок".
Методы исследования: электромиография
Выявление спинальной амиотрофии базируется (кроме раннего проявления и типичной клинической картины) на результатах ряда дополнительных исследований. Из них стоит выделить электромиографию. Практически во всех случаях обнаруживается биоэлектрическая спонтанная активность в состоянии покоя при наличии потенциалов фасцикуляций. На фоне произвольных сокращений констатируется электрическая активность уреженного характера с ритмом "частокола". Это указывает на увеличение продолжительности потенциала и явления синхронизации.

Патоморфологическое исследование
Оно позволяет выявить уменьшение числа клеток передних спинномозговых рогов, а также изменения дегенеративного типа. Патологические нарушения резко выражены в районе шейного и поясничного утолщений, в двигательных ядрах черепных нервов. Обнаруживаются также изменения передних корешков, в интрамускулярных зонах нервных окончаний. Отмечается исчезновение и излишнее разветвление нормальных терминал.
Биохимический анализ
Это исследование позволяет выявить изменения углеводного обмена. Так, было обнаружено, что при спинальной амиотрофии гликолиз у пациентов приближен к эмбриональному типу. Достаточно часто выявляются существенные изменения креатин-креатининового обмена - повышение экскреции креатина, снижение выделения креатинина. Необходимо также отметить, что концентрация ферментов в сыворотке крови практически неизменна.

Спинальная амиотрофия: лечение
Терапия патологии сводится к назначению ЛФК и массажа. Эти процедуры должны выполняться регулярно. Радикальные методы лечения отсутствуют. В определенной степени облегчение может принести прием ряда медикаментов. В частности, специалисты рекомендуют такие средства, как "Сангвинарин", "Галантамин", "Оксазил", "Прозерин". Дополнительно назначаются витамины группы В. При выраженных проявлениях заболевания может быть рекомендовано повторное переливание крови в малых дозах.
Амиотрофия – болезнь, описанная учеными в конце 19 века и охарактеризованная изменениями определенных групп мышц, нервов на периферии и самого спинного мозга. В научных трудах было подмечена атрофическая симметричность клеток передних корешков и рогов спинного мозга. Позже выделили форму заболевания более легкую, где поражаются только клетки передних рогов спинного мозга, назвали ее – нозологической. Спинальная мышечная атрофия – наследственное генетическое заболевание. Происходит потеря мышечной активности за счет деградации нейронов передних рогов спинного мозга. Страдает поперечнополосатая мускулатура нижнего пояса, шеи и головы, верхний плечевой пояс задействован в меньшей степени. У человека наблюдаются нарушения самопроизвольных движений, например, ползанья у ребенка или ходьбы. Но психически человек не страдает, остается чувствительность при спинальной атрофии.
В чем причина? Заболевание развивается довольно редко, так как передается по аутосомно-рецессивному типу. Это значит, что у обоих родителей должно быть изменение генетического материала в пятой хромосоме (SMN), тогда скорее всего (но не 100%), родиться ребенок с таким заболеванием. В процессе этого сбоя происходит уменьшение выработки белка SMN, который и ведет к потере моторных нейронов.
Если же человек является носителем такой мутации, то никаким образом на его здоровье это не влияет.
Имеется классификация данного заболевания. Деление происходит по возрастным характеристикам, сложности протекания и возрастных особенностей:
- Спинальная мышечная атрофия 1 типа или младенческий (сма 1), так же можно встретить название: болезнь Вердинга–Гоффмана. Страдают дети до первого полугода жизни. Часто исход смертельный. У новорождённых возникают стойкие нарушения сосательного рефлекса, а также глотания.
- Атрофия 2 типа или промежуточный (сма 2) или болезнь Дубовица. Болеют дети от 7 месяцев до 1 года. При этой форме дети могут питаться и глотать, но самостоятельно ходить или сидеть не могут. К сожалению, погибают дети из-за сопутствующих заболеваний, например, от застойных пневмоний из-за малоподвижности.
- 3 тип или юношеский (ювенильный) (сма) 3 или болезнь Кюгельберга – Веландера. Заболевание начинается с 1,5 лет и может быть даже у взрослых. Такие пациенты могут самостоятельно стоять, но передвигаются в инвалидном кресле.
- 4 тип или взрослый – (сма 4) или взрослая форма. Проявляется с 35 лет, очень медленно развивается, но может привести к полной потере двигательной активности.
Клиническая картина
Заболевание делиться на 4 типа. Тяжесть болезни и ее исход зависит от сложности течения заболевания от возраста пациента. Как правило, в любой форме ставят инвалидность. В тяжелых типах болезнь может требовать постоянной медицинской помощи. При любом из видов СМА не страдает чувствительность, так как в процессе не участвуют чувствительные нервные волокна. Интеллектуальная сторона так же не задействована, поэтому ребенок легко обучаем на равне со своими сверстниками. А вот сердечная и дыхательная системы берут на себя весь удар последствий этой болезни. Смерть наступает в основном из-за затяжных застойных пневмоний или бронхитов, в следствии практически отсутствующей двигательной активности.
Тип 1. Болезнь Верднига-Гоффмана

Спинально-мышечная атрофия Верднига-Гоффмана начинает проявляться еще во внутриутробном развитии плода. Заболевание получило название по фамилиям ученых, Верднигом и Гофманом были описаны одни из первых морфологических признаков болезни. Примерно с двадцать восьмой недели беременности наблюдается слабая активность плода. После рождения у ребенка начинают нарастать определенные признаки этого заболевания в первые пол года жизни. Ребенок постоянно лежит, практически обездвижено, не переворачивается, наблюдается тик пораженных мышц, ножки не сгибает.
В результате развития болезни 1 типа происходит атрофия мышц, они значительно уменьшаются. Глотка, диафрагма, межреберные и грудные мышцы не снабжается нервными клетками, что затрудняет процесс глотания. Это все ведет к застойным пневмониям, их обязательно нужно своевременно диагностировать, в противном же случае это приведёт к летальности. Еще одна характерная черта при заболевании Верднига Гоффмана – деформация скелета, из-за ослабленности мышц, которые не способны удерживать скелет.
Если ребенок пытается сидеть, то при заболевании верднига, как правило, формируется сколиоз. Уплощается так же грудная клетка, это ведет к затруднению дыхания и нарушению функций сердечно – сосудистой системы.
Тип 2. Младенческая форма

Этот тип заболевания диагностируется позже, примерно с полутора лет, как только ребенок предпринимает попытки самостоятельно ползать или сидеть. Симптомы заболевания у детей замечают уже при рождении. Первые признаки – это задержка физического развития, очень вялая двигательная активность, сухожильные рефлексы снижены. Эта форма заболевания переносится легче, чем тип верднига. Атрофия мышц протекать медленно, и ребенок может проживать до 18 лет. Пациент может самостоятельно обслуживать себя в последствии, так же сидеть, стоять, есть, но передвигаться только с помощью инвалидного кресла. За счет малой двигательной активности, могут присоединятся различные респираторные инфекции, а также воспаление легких, в самых тяжелых формах их проявлений.
Тип 3. Ювенильный или болезнь Куленберга – Веландера
Характеризуется развитием у детей с 2 лет и старше. Медленное развитие болезни, но прогрессирующее. Направлена на угнетение физической активности. Изначально ребенок двигается, ходит, поднимается и спускается по лестнице, а со временем двигательная активность затрудняется. Это приводит так же спинально-мышечной атрофии. В первую очередь страдает мышечная система ног, позже спины, шеи и в конце верхнего плечевого пояса. Самоуход возможен довольно длительное время, но в любом случае этот тип так же ведет к инвалидности.
Тип 4. Взрослая форма заболевания

Встречается редко и начинает свое развитие с 30 лет жизни человека. В основном задействованы шейные и головные мышцы, пациент может полностью себя обслуживать. Признаками спинально-мышечной атрофии может быть слабая мимическая деятельность мышц лица, подёргивание языка, некое ограничение подвижности головы. Как правило, это считается благоприятным исходом заболевания.
Диагностика
Диагностируют по совокупности первых признаков заболевания. Для подтверждения диагноза у пациента берут биопсию потенциально пораженной мышцы, лабораторными методами уточняют патологические изменения. Для того, чтобы обнаружить патологию у ребенка или взрослого назначают МРТ и КТ.
Анализ ТМС широко используется в диагностике заболевания, в при генетическом скрининге потенциально подозреваемых больных. ТМС – транскраниальная магнитная стимуляция, диагностический амбулаторный метод, основанный на стимуляции коры головного мозга с помощью коротких магнитных импульсов, не приносит каких-либо болезненных ощущений.
Профилактика заключается в беседе с врачом-генетиком пары, у которой это заболевание уже имеется в семейном анамнезе или имеется ген СМА. Составляется генеалогическое древо и делается вывод о процентном соотношении и риске передачи наследственного заболевания ребенку. Так же проводится биопсия ворсин хориона. Но ни один метод диагностики или профилактики не дает 100 % положительный результат о наличии генетического заболевания у детей.
Лечение

Медицина еще не изобрела средство лечения спинально-мышечной атрофии. Вся суть лечение направленна на поддержание жизнедеятельности больного и избежание каких-либо осложнений. Что входит в совокупность мер по лечению заболевания:
- Лекарственная терапия, при которой нервный импульс будет лучше проходить от центрального отдела нервной системы к периферическим отделам. Так же лекарственная терапия должна включать препараты с содержанием калия.
- Витаминотерапия, обязательно комплексная. Группы В.
- Препараты ноотропной терапии, для восстановления кровоснабжения в нервной ткани.
- Физиологические процедуры, как закрепление вышеперечисленной терапии, например, парафиновые ванны.
- Обязательным является массаж при спинально мышечной атрофии, для мышечного тонуса.
- Для укрепления связок используется лечебно – профилактическая физкультура.
В самых тяжелых формах, требуется реанимационная мероприятия.
Многие ученые и медики работают над таким препаратом, который бы восполнял определённый белок при этом заболевании. Это дает надежду на полное выздоровление от тяжелой генетической болезни, пусть даже искусственным путем и пожизненным употреблением лекарственных средств.
Спинальной амиотрофией Верднига-Гоффмана называется наследственная патология нервов, которая поражает тот их участок, что контролирует скелетные мышцы. Ее характеризует слабость сразу всех мышц в организме. Более половины нервных клеток, которые контролируют мышцы, располагаются в спинном мозге. Именно потому заболевание названо «спинальным». «Мышечным» оно также названо из-за пагубного влияния на мышцы: те не получают от этих самых нервов никаких сигналов. «Атрофия» является медицинским термином, который обозначает истощение или усыхание того, что не используется. Здесь это касается недействующих мышц. Человек с подобной болезнью не имеет никакой возможности ни сидеть, ни передвигаться, ни даже обслуживать себя. Вылечить это невозможно. Проведя дородовую диагностику, можно предотвратить рождение малыша с таким заболеванием. Здесь мы поговорим, как эта патология наследуется, каковы ее проявления, а также о том, как можно помочь больному человеку.
Спинальная амиотрофия Верднига-Гоффмана названа в честь двоих ученых, первыми ее описавших. Во второй половине XIX столетия они доказали морфологическую сущность болезни. Сначала оба ученых считали, что у данной патологии только одна форма. Но уже в XX столетии ученые Кукельберг и Веландер открыли другую ее клиническую форму, с генетической причиной, схожей с той, которая и у открытой Верднигом и Гоффманом. Сейчас известно несколько клинических форм спинальной амиотрофии. Их объединяет общий наследственный дефект.
Эта патология наследственная. Она основана на мутации в пятой хромосоме. Мутирует тот ген, благодаря которому синтезируется белок SMN. Данный белок необходим для того, чтобы двигательные нейроны развивались именно так, как нужно. Стоит пятой хромосоме мутировать, и это негативно скажется на двигательных нейронах, помешав их развитию, а то и вовсе их разрушив. В результате мышца не может получать управляющие сигналы от нервов, а значит, не может и функционировать. Получается, не выполняется ни одно связанное с ней движение.

У мутировавшего гена аутосомно-рецессивный тип наследования. Фраза расшифровывается так: для развития спинальной амиотрофии нужно, чтобы у обоих родителей был мутантный ген. Если проще, то заболевание не разовьется, если хоть один из родителей не был носителем мутировавшего гена. В то же время они сами не болеют: у людей гены парные, а у отца и у матери ребенка доминирует здоровый ген. В таком случае больной малыш рождается примерно в четверти случаев. Ученые подсчитали, что около 2% живующих людей - носители гена с такой мутацией.
Классификация
Известны три разновидности данной патологии.
- Самая тяжелая, проявляющаяся раньше остальных.
- Среднетяжелая.
- Самая легкая, проявляющаяся в самом позднем возрасте.
По мнению некоторых врачей, существует еще одна разновидность: умеренная/мягкая СМА, которая проявляется уже у взрослого человека.
Стоит отметить, что кроме спинальной амиотрофии Верднига-Гоффмана существуют и другие типы СМА, которые отличаются симптомами и типами наследования. Они указаны в таблице ниже.
Таблица №1. Виды СМА.
| Название CMA | Тип наследования | Особенности и симптомы |
|---|---|---|
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном y пожилых, поражает бульбарные нервы чepeпa, вызывает нисходящий паралич. |
| SMAХ2 | Х - сцепл. рецессивный | Врождённая агрессивная форма, приводящая к смерти до 3 - х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х - сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов. |
| Дистальная ДCMA1 | Аутосомно - рецессивный | Врождённая, поражаются в основном руки, возможны тяжелее дыхательные нарушения. |
| Дистальные формы ДCMA2 - ДCMA5 | Аутосомно - рецессивный | Все четыре формы отличаются медленным прогрессированием, ДCMA5 диагностируется y молодых. |
| Ювенильная SMA (тип HMN1) | Аутосомно-доминантный | Встречается в юности |
| Врождённая спинальная aмиoтpoфия | Аутосомно-доминантный | Нарушение иннервации и атрофия бедер, стоп, коленей c контрактурой и деформацией; иногда поражается голосовые связки. |
| SMA Финкeля | Аутосомно-доминантный | Начинается преимущественно в 35 - 37 лет, но зафиксированы случаи заболевания и в детском возрасте. Медленно развивается вначале в ногах, a затем в руках. Активность и рефлексы снижены, наблюдается непроизвольное дрожание (фасцикуляция). |
| SMA (тип LED1) | Аутосомно-доминантный | Атрофия нижних конечностей y новорождённых. |
| CMA c вpoждeнными кocтными пepeлoмaми | Аyтocoмнo-peцeccивный | Тяжелее симптомы, как про бoлeзни. Вepднигa-Гoффмaнa, отягощённые переломами. |
| CMA c гипоплазией | Аутосомно-доминантный | Врождённая аномалия головного мозга c церебральными симптомами, микроцефалией и задержкой развития. |
Симптомы патологии
Врожденная разновидность заболевания (СМА I) начинает проявляться до того, как малышу исполнится полгода. До этого такой ребенок может вяло двигаться. Не так уж редко спинальную амиотрофию у малыша можно заметить еще в самом начале постэмбрионального периода его жизни - у него угасают глубокие рефлексы:
- у малыша недостаточно громкий крик;
- ему трудно сосать;
- он не может держать головку.

Определить такую патологию у младенца можно с первых дней жизни
Если подобное становится заметно позже, что происходит редко, малыш может научиться держать головку, а то и сидеть, однако патология быстро сводит такие умения к нулю. Характерно и следующее:
- ранние проблемы с речью;
- ухудшение глоточного рефлекса;
- фасцикулярные подергивания языка.
Этот вид патологии может прилагаться к олигофрении, а также к патологиям развития скелета:
- деформациям грудной клетки (воронкообразной/килевидной ее форме);
- искривлению позвоночника (сколиозу);
- суставным контрактурам.
Нередки и другие врожденные заболевания. Например:
- гемангиомы;
- гидроцефалия;
- косолапость;
- дисплазия тазобедренных суставов;
- крипторхизм.
СМА I самая «вредная»: ее сопровождают развивающийся паралич, а также парез мускулатуры, отвечающей за дыхание. Из-за последнего развивается дыхательная недостаточность, из-за которой больной даже может погибнуть. А нарушение глотания может оказаться причиной попадания пищи в дыхательные пути и развития аспирационной пневмонии. Она тоже может привести к смерти.
СМА II начинает проявляться после того, как малышу исполняется полгода. В таком возрасте малыш уже удовлетворительно развит, он может стоять, держать головку, садиться, переворачиваться. Однако почти никогда больной малыш не успевает овладеть навыками ходьбы. В большей части случает данное заболевание проявляется после того, как малыш перенесет какую-либо острую инфекционную патологию. Например, пищевую токсикоинфекцию.
Когда СМА II только начинает проявляться, у малыша в ножках появляются периферические парезы, которые потом быстро оказываются в ручках, в туловище. Проявляется диффузная гипотония мышц, начинают исчезать глубокие рефлексы. Можно заметить и следующее:
- контрактуры сухожилий;
- тремор пальцев;
- фасцикуляции (непроизвольное дерганье) языка.
Важно! Позже начинает проявляться бульбарный синдром, развивается дыхательная недостаточность. Благодаря замедленному, если сравнивать с врожденной формой, развитию этой разновидности СМА, больной человек обычно доживает до пятнадцати лет.
СМА III , она же амиотрофия Кугельберга-Веландера - наименее вредная разновидность спинальной амиотрофии. Начинает проявляться, когда ребенку исполняется два года. Иногда может протекать бессимптомно даже до 30 лет. Впрочем, в этом случае пациент уже далеко не ребенок, что, однако, не делает его здоровее. При СМА III не задерживается развитие психики, пациент долго может двигаться без посторонней помощи. У него даже есть шансы и в глубокой старости иметь возможность самостоятельно себя обслуживать.
СМА IV , она же взрослая форма спинальной амиотрофии, представляет собой медленно развивающуюся патологию. Она обычно начинается после того, как человеку исполняется 35 лет. Такая форма если и сокращает жизнь, то незначительно. С другой стороны, у больного наблюдаются следующая картина:
- слабые проксимальные мышцы;
- фасцикуляции;
- ухудшение сухожильных рефлексов;
- потеря способности к ходьбе.

Электромиограмма показывает «ритм частокола» - спонтанную ритмическую активность. Так можно выявить патологию передних спинномозговых рогов. Если провести морфологическое исследование биоптатов мышц, можно заметить атро- и гипертрофированные волокна первого и второго типов. Также скапливаются мелкие круглые волокна, которые перемежаются с волокнами гипертрофированными - это «пучковая» атрофия. Если провести патоморфологическое исследование, становится заметно набухание/сморщивание/атрофия мотонейронов спинномозговых передних рогов, а довольно часто - еще и ядер нервов, которые выходят из головного мозга.
Как диагностируют данную патологию?
Чтобы поставить правильный диагноз, невролог должен иметь информацию о том, в каком возрасте у человека появились первые симптомы, как быстро они прогрессируют. Также он должен выяснить неврологические данные. Причем здесь важнее всего информация о том, нет ли у пациента периферических двигательных нарушений вместе с полным сохранением чувствительности. Еще неврологу важно узнать, не имеет ли больной врожденных отклонений, костной деформации. Амиотрофию Верднига-Гоффмана может диагностировать и неонатолог. При помощи дифференциальной методики можно диагностировать:
- миопатию;
- развивающаяся мышечная дистрофия Дюшенна;
- боковой амиотрофический склероз;
- сирингомиелия;
- полиомиелит;
- синдром вялого ребенка;
- обменные патологии.
Чтобы подтвердить результаты диагностики, делают электронейромиографию. Так называется обследование нервно-мышечного аппарата. Оно позволяет обнаружить изменения, нужные, чтобы исключить первично-мышечный вид заболевания, а также убедиться в наличии болезни мотонейрона. Биохимического анализа крови слишком мало, чтобы выявить сколь-либо серьезное повышение уровня креатинфоскиназы, которое бывает во время развития дистрофии мышц. Сделав МРТ/КТ позвоночника, изредка можно обнаружить атрофию передних рогов спинного мозга, однако при помощи этой методики можно убедиться в том, что другого спинального заболевания нет.

Окончательно ставится диагноз «амиотрофия Верднига-Гоффмана» только когда будут получены данные биопсии мышц, а также анализа ДНК. При помощи морфологического обследования мышечного биоптата можно выявить патогномоничную пучковую атрофию мышц, перемежающуюся зонами атрофии миофибрилл и здоровой ткани, отдельными гипертрофированными миофибриллами, а также местами соединительных разрастаний. В обязательные генетические исследования входит прямая и косвенная диагностика. Прямая методика позволяет выявить гетерозиготное носительство генной аберрации, а это необходимо при генетическом консультировании родственников пациентов, а также супругов, планирующих рождение ребенка. Во всем этом важно количественное исследование генов локуса СМА.
Проведя дородовую генетическую экспертизу, можно понизить риск появления на свет малыша, болеющего амиотрофией Верднига-Гоффмана. Чтобы получить генетический материал плода, следует применять инвазивные методики диагностики плода:
- амниоцентез;
- биопсию хориона;
- кордоцентез.

Обнаружив у плода амиотрофию Верднига-Гоффмана, следует поставить вопрос об аборте.
Дифференциальная методика диагностирования заболевания
По симптомам данную патологию можно перепутать с врожденной миопатией. Это плохой мышечный тонус. Для исключения мышечной гипотонии используется биопсия. На спинальную амиотрофию Верднига-Гоффмана похож острый полиомиелит. У этой патологии бурное, сопровождающееся резко повышенной температурой тела и множественными асимметричными параличами, начало. Через несколько суток острая фаза полиомиелита переходит в восстановительную. При гликогенозах, а также врожденных миопатиях тоже понижается тонус мышц.

Но эти патологии отличаются от спинальной амиотрофии тем, что их провоцируют не мутации гена, а следующие факторы:
- проблемы с метаболизмом;
- карцинома;
- гормональное нарушение.
При этом необходимо исключить и такое:
- болезнь Гоше;
- синдром Дауна;
- ботулизм.
Каким образом лечат данную патологию, а также как спрогнозировать ее дальнейшее протекание
Сегодня еще не существует этиопатогенетического лечения спинальной амиотрофии. Сейчас ее лечат, улучшая обмен веществ в периферической нервной системе, а также в мышцах, чтобы просто задержать развитие симптомов.
При этом в разных сочетаниях используются средства следующих групп:
- нейрометаболиты - лекарства, производимые из гидролизата свиных мозгов, гамма-аминомасляная кислота, а также пирацетам;
- средства, облегчающие передачу импульсов к мышцам - галантамин, сангвинарин, неостигмин, ипидакрин;
- медикаменты, улучшающие трофику миофибрилл - коэнзим Q10, L-карнитин, Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, Карнитина хлорид, Метионин, Калия оротат, Токоферола ацетат;
- витамины В - Мильгамма, Нейровитан, Комбилипен;
- анаболики - Ретаболил, Неробол;
- лекарства для улучшения проводимости импульсов между нервами и мышцами - Прозерин, Нейромидин, Дибазол, Галантамин;
- медикаменты для того, чтобы улучшить кровообращение - никотиновая кислота, скополамин.

Кроме того, при спинальной амиотрофии полезно делать ЛФК, а также проводить сеансы мягкого массажа.
Сегодняшние технологии являются большим подспорьем для облегчения жизни больных, а также их близких. В этом им помогают портативные аппараты ИВЛ, а также автоматизированные инвалидные коляски. Для улучшения подвижности больных существуют разнообразные методики ортопедической коррекции. А вот главные перспективы лечения СМА заключаются в непрерывно развивающейся генетике, а также в поиске возможностей использования генной инженерии для корректирования генетических заболеваний.
Анестезия
Если человеку вне зависимости от его возраста требуется операция, необходимо следовать определенным мерам предосторожности. Вся хирургическая бригада обязана быть в курсе того, что представляет собой СМА. Больше всего это касается анестезиолога.
В начале развития данной патологии в клетках мышц, не получающих нервных сигналов, порой возникают аномалии, обусловленные попытками мышц «дотянуться» до связанных с ними нервов. Из-за таких аномалий миорелаксанты, нередко используемые при хирургическом вмешательстве, могут оказаться опасны. Имея представление о подобных проблемах, можно подобрать безопасные препараты.
Диета при спинальной амиотрофии
На данный момент еще не подтверждено, что какая-либо диета приносит пользу при СМА.
По мнению большого количества родителей, диета, включающая в себя много белка или специальные добавки к пище, могут повысить силу мышц ребенка. Но, несмотря на очевидную необходимость хорошего питания для больного ребенка, еще не доказано, что ему нужен именно определенный рацион. Причем некоторые продукты даже могут нанести вред его организму.
К примеру, аминокислотное меню порой чревато еще большими проблемами у тех детей, у которых в организме слишком мало мышечной ткани. По мнению некоторых специалистов, при недостатке мышечной ткани, та не может правильно перерабатывать аминокислоты и тогда их уровень в крови повышается слишком сильно.

Некоторым детям полезнее питаться понемногу, причем чаще трех-четырех раз в день. Нужно просто разделить для больного все количество пищи, принимаемое здоровым сверстником больного за день, на несколько частей.
Среди больных СМА есть обладатели лишнего веса. Возможно, что причина этого кроется в слишком калорийном рационе вкупе с недостатком движения. Если есть возможность, больной должен, заручившись помощью врача и диетолога, привести свой вес в норму. Это важно не только для здоровья и внешности, но и для тех, кто ухаживает за таким больным. Ведь они каждый день помогают больному подниматься и перемещаться.
Важно! Есть врачи, советующие принимать отпускаемые без рецептов пищевые добавки. Например, креатин, коэнзим Q10. Сейчас ведутся исследования того, как креатин действует при данной патологии.
ЛФК
По мнению большинства врачей, комфортная физическая активность, если только не впадать в крайности, весьма полезна для самочувствия и здоровья больного СМА.
Суставам необходимо обеспечивать подвижность, а не подвергать их риску повреждений. При этом следует сохранять объем движений, чтобы сохранять суставы эластичными. При этом нужно поддерживать циркуляцию крови. Также, что важнее всего для детей, надо поддерживать мобильность высокой, чтобы исследовать окружающее.
Важно! Лучше всего проводить занятия в бассейне, наполненном водой, имеющей температуру 30-32 градуса тепла. Но, во-первых, человек, больной СМА, не должен заниматься плаванием сам, а во-вторых, нужно соблюдать определенные меры безопасности.
По мнению некоторых врачей, уделять массу внимания постепенному понижению в организме и без того недостаточного числа мотонейронов не так уж и нужно. Для определения, следует ли это учитывать, разрабатывая комплекс ЛФК, нужны исследования. Мнения профессионалов расходятся: одни считают, что перегрузить организм упражнениями невозможно, другие же — что, делая гимнастический комплекс «до посинения», можно форсировать отмирание оставшихся мотонейронов. Так что при ЛФК требуется аккуратность и гимнастику необходимо прекратить, не доводя себя до изнеможения.
Программы трудо- и физиотерапии полезны для людей любого возраста, если те хотят научиться максимально рационально использовать оставшиеся мышечные функции, а также разобраться, как лучше всего справляться с повседневными задачами.
Сейчас можно найти приспособления, которые полезны даже малышам – для изучения окружающего пространства. Маленьким детям может помочь все, что только придумал для решения этой задачи человеческий гений, начиная от ходунков и заканчивая ортезами.
Более того, есть семьи, которые самостоятельно придумывают и изготавливают свои приспособления, снабженные особыми функциями. К примеру, такие, у которых можно менять высоту, чтобы ребенок мог хоть ползать на полу, хоть сидеть на столе.
Человеку любого возраста, если у него СМА, ощутимо полезны приспособления для решения повседневных задач, не вызывающих затруднений у здоровых людей.
Способ предупреждения данной патологии
Для предупреждения амиотрофии Верднига-Гоффмана у новорожденного родители должны вовремя пройти диагностику генетических отклонений, а также, вынашивая ребенка, будущей матери следует провести еще и претональную ДНК-диагностику малыша. Если у того выявляется амиотрофия Верднига-Гоффмана - встает вопрос об аборте.

Как спрогнозировать развитие патологии
Сейчас спинальная амиотрофия Верднига-Гоффмана неизлечима. Прогнозы абсолютно неблагоприятны. Если такая проявляется у малыша в первые дни после его рождения, тот обычно умирает до того, как ему исполнится полгода. Если же симптомы начинают проявляться после трехмесячного возраста, средний возраст такого ребенка равняется паре лет. Максимальный возраст, до которого может дожить такой больной, равен восьми годам. Ранний детский тип этой патологии прогрессирует куда медленнее, и такой ребенок доживает до подросткового возраста - четырнадцати-пятнадцати лет.
Это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
МКБ-10
G12.0 Детская спинальная мышечная атрофия, I тип [Верднига-Гоффмана]

Общие сведения
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию - амиотрофию Кугельберга-Веландера . Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины
Амиотрофия Верднига-Гоффмана - наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) - ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии
Врожденная форма (СМА I) клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов . У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия , косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности , выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии , которая может явиться смертельно опасным осложнением спинальной амиотрофии .
Ранняя детская форма (СМА II) дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера (СМА III) - наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития , длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Диагностика
В диагностическом плане для детского невролога имеет значение возраст появления первых симптомов и динамика их развития, данные неврологического статуса (в первую очередь наличие двигательных нарушений периферического типа на фоне абсолютно сохранной чувствительности), наличие сопутствующих врожденных аномалий и костных деформаций. Врожденная амиотрофия Верднига-Гоффмана может быть диагностирована неонатологом. Дифференциальный диагноз проводится с миопатиями , прогрессирующей мышечной дистрофией Дюшенна, боковым амиотрофическим склерозом , сирингомиелией, полиомиелитом, синдромом вялого ребенка , ДЦП , обменными заболеваниями.
С целью подтверждения диагноза проводится электронейромиография - исследование нервно-мышечного аппарата, благодаря которому выявляются характерные изменения, исключающие первично мышечный тип поражения и указывающие на патологию мотонейрона. Биохимический анализ крови не выявляет существенного повышения креатинфосфокиназы, характерного для прогрессирующей мышечной дистрофии. МРТ или КТ позвоночника в редких случаях визуализируют атрофические изменения передних рогов спинного мозга, но позволяют исключить другую спинальную патологию (гематомиелию , миелит , кисту и опухоль спинного мозга).
Окончательный диагноз «амиотрофия Верднига-Гоффмана» устанавливается после получения данных биопсии мышц и генетических исследований. Морфологическое изучение мышечного биоптата выявляет патогномоничную пучковую атрофию мышечных волокон с чередованием зон атрофии миофибрилл и неизмененной мышечной ткани, наличием отдельных гипертрофированных миофибрилл, участков соединительнотканных разрастаний. Проводимый генетиками ДНК-анализ включает прямую и косвенную диагностику. С помощью прямого метода возможна также диагностика гетерозиготного носительства генной аберрации, что имеет важное значение в генетическом консультировании сибсов (братьев и сестер) больных лиц, супружеских пар, планирующих беременность. При этом большую роль играет количественный анализ числа генов локуса СМА.
Дородовый анализ ДНК позволяет снизить вероятность рождения ребенка с болезнью Верднига-Гоффмана. Однако для получения ДНК-материала плода необходимо использовать инвазивные методы пренатальной диагностики: амниоцентез , биопсию хориона, кордоцентез . Амиотрофия Верднига-Гоффмана, диагностированная внутриутробно, выступает показанием к искусственному прерыванию беременности .
Лечение амиотрофии Верднига-Гоффмана
Этиопатогенетическая терапия не разработана. В настоящее время амиотрофия Верднига-Гоффмана лечится путем улучшения метаболизма периферической нервной системы и мышечной ткани с целью замедлить прогрессирование симптомов. В терапии применяют комбинации препаратов различных фармакологических групп: нейрометаболиты (препараты на основе гидролизата головного мозга свиньи, витамины гр. В, гамма-аминомасляная кислота, пирацетам), облегчающие нервно-мышечную передачу (галантамин, сангвинарин, неостигмин, ипидакрин), улучшающие трофику миофибрилл (глютаминовая к-та, коэнзим Q10, L-карнитин, метионин), улучшающие кровообращение (никотиновая к-та, скополамин). Рекомендована лечебная физкультура и детский массаж .
Современное развитие техники позволило несколько облегчить жизнь пациентов и их родственников, благодаря применению автоматизированных инвалидных колясок и портативных аппаратов ИВЛ. Улучшить подвижность пациентов помогают различные методы ортопедической коррекции. Однако основные перспективы в лечении СМА связаны с развитием генетики и поисками возможностей коррекции генетических аберраций методами генной инженерии.
Прогноз
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда - к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.